

 Pour de nombreux sites, la constitution du dossier de la CCS passe a minima par la mise en cohérence de documents existants, mais aussi par la nécessité de réinterroger les analyses et les pratiques existantes.
Pour de nombreux sites, la constitution du dossier de la CCS passe a minima par la mise en cohérence de documents existants, mais aussi par la nécessité de réinterroger les analyses et les pratiques existantes.
L'esprit de la stratégie de maîtrise de la contamination (CCS) et de la nouvelle annexe n’est pas focalisé sur la recherche de la conformité, mais bel et bien sur une approche globale dans la gestion de la maîtrise des contaminations. En tout premier lieu, il s’agit bien d’une volonté de maîtrise globale, et non d’une liste d’actions et de contrôles à mettre en place.
L'idée est, pour un industriel assurant la fabrication de produits médicamenteux stériles, de produire un « document chapeau » officiel, définissant globalement et de manière synthétique et cohérente la stratégie mise en place destinée à être présentée lors des inspections. En ligne de fond, il s'agit d'assurer le lien entre toutes les procédures et pratiques mises en place sur le site de production pour un procédé de fabrication donné, et de savoir justifier tous les choix réalisés, tant en termes de conception que de méthodes, de contrôle, d'exploitation des résultats… bref, de savoir en permanence répondre à la question « Pourquoi ? ».
Concrètement, et en cohérence avec d'autres référentiels déjà applicables dans d'autres pays au niveau réglementaire (les exigences de la
Préalablement à la CCS auront été élaborées une définition précise des besoins des utilisateurs et du laboratoire en termes de production et de maîtrise des contaminations (particules inertes, micro-organismes, endotoxines) ainsi que des évaluations des risques documentées, précises, permettant de justifier toutes les actions et décisions mises en place sur le site dans un procédé de fabrication.
On pourrait donc penser qu'une stratégie de maîtrise de la contamination consiste simplement à mettre en place des contrôles particulaires, microbiologiques et autres prélèvements, et d'en exploiter les résultats. En réalité, cette stratégie recouvre un champ bien plus large et nécessite la maîtrise de nombreux paramètres… C'est précisément ce qui est exigé par la réglementation pharmaceutique avec la nouvelle annexe 1.
Attendus de la CCS
Rappelons les attendus de la CCS :
- définir tous les points de contrôle critiques et évaluer l'efficacité de tous les contrôles (conception, procédures, aspects techniques et organisationnels) et des mesures de surveillance employées pour gérer les risques pour la qualité et la sécurité des médicaments ;
- établir une assurance solide de la prévention de la contamination ;
- conduire à une amélioration continue des méthodes de fabrication et de contrôle, évaluée et mise à jour régulièrement ;
- mentionner les contrôles existants, et une série d'événements et de mesures interdépendants pour une efficacité collective.
Quelques considérations concernant l'analyse de risque
Elle s'appuie sur les besoins utilisateurs formulés, ainsi que les attentes réglementaires de l'annexe 1 : ne pas omettre dans l'analyse de risque de considérer le respect des exigences réglementaires spécifiques décrites et de les évaluer en termes de gravité (HSE, patient, ou production).
Pour la rédaction de la CCS
La démarche à suivre consiste à décrire dans un document les moyens de maîtrise à utiliser, en s'assurant que les exigences ont été intégrées dans les pratiques et procédures opérationnelles du site. Concrètement, les éléments suivants doivent être pris en compte, et la CCS doit faire référence aux documents internes en place pour maîtriser chacun des points suivants.
Conception de l'usine et des procédés
Pour la conception de l'usine, une réflexion globale se traduisant par une description précise intégrant les flux personnels, les flux matériels et équipements, les flux produits, les flux relatifs aux déchets de production et l'aéraulique dans l'agencement général des lieux est attendue. Ainsi doivent y figurer des plans décrivant les classifications des environnements, leur rationnel, les principes des dispositifs de traitement et de diffusion de l'air (notamment PID des CTA, niveaux de filtration choisis, types de matériaux et de gaines…), les gradients de pression spécifiés, ainsi que toutes les données relatives aux choix de matériels et technologies associés. Cela inclut également les informations relatives aux technologies aseptiques choisies, aux systèmes de contrôle de la contamination (ex. : dispositifs de monitoring), l'articulation des sas… Comme évoqué dans le texte, il est nécessaire, le cas échéant, de justifier de la non-utilisation de certaines technologies avancées comme les technologies barrières.
Locaux et équipements
Le listing des locaux, des équipements et des systèmes doit être établi ainsi que leurs niveaux de criticité ; en effet, le plan directeur de validation permet de définir une stratégie et les éléments clés du programme des qualifications à mener, les efforts de qualification associés (même si l'annexe 1 impose bon nombre d'exigences en termes de qualification), pour prouver que ces systèmes fonctionnent de manière fiable et reproductible.
Personnel
Une description du processus de formation précisant comment le personnel est formé, évalué et habilité pour travailler dans la zone aseptique est nécessaire : il comprend généralement une formation théorique (incluant le visionnage de visualisations de flux d'air pour les personnels intervenant en salle propre), une formation pratique incluant l'habillage (la prise de connaissance des procédures d'habillage, les tests et prélèvements réalisés sur le personnel pour valider leur méthodes d'habillage…), la participation aux opérations de remplissage aseptique et aux

- Le travail imposé par la CCS doit pousser les gestionnaires à sortir des logiques de recherche de conformité pour porter leur attention sur l'efficacité, la cohérence et la pertinence des mesures de prévention de la contamination.© Novartis
Utilités
Une description des systèmes de distribution d'eau, de vapeur, de gaz comprimés et les autres utilités critiques des installations est à fournir, en détaillant la conception, les essais de qualification, le régime d'échantillonnage pour les contrôles de routine, les emplacements justifiés des points de prélèvement, les techniques d'échantillonnage choisies et leur périodicité, les critères d'acceptation en cours de fabrication, les spécifications et résultats attendus pour chaque système.
Contrôles des matières premières et contrôles en cours de fabrication
La maîtrise des matières premières et des contaminations nécessite une description des spécifications pour les matériaux achetés, répondant aux exigences de qualité pharmaceutique adéquates. Une description des choix des méthodes d'essais et d'échantillonnage de routine est à fournir pour contrôler la qualité des matières premières. Des contrôles microbiologiques à périodicité renforcée sont à définir et à mettre en œuvre en routine.
Contenants et produits clos
Les spécifications pour les matériaux achetés doivent être définies et justifiées. La fourniture de résultats de contrôles microbiologiques relatifs aux contenants et produits de la part des fournisseurs s'avère pertinente. De plus, les pratiques des fournisseurs de composants ayant subi des opérations de nettoyage et/ou stérilisés en amont doivent être validées et décrites dans la CCS.
Qualification des fournisseurs de composants et services critiques
La sélection et la gestion efficace de ces fournisseurs doivent être démontrées avec une description des pratiques en matière de sélection initiale des fournisseurs (par exemple avec des grilles de notation multicritères) et une politique définissant leur évaluation, les périodicités des audits et le suivi des fournisseurs. Cette approche fondée sur les risques doit idéalement être décrite dans des documents référencés. Les mécanismes de décision lors des achats doivent suivre une logique similaire, et des processus de décision clairement définis. En effet, comment s'assurer qu'un matériau de qualité pharmaceutique ne sera pas remplacé par un matériau plus basique choisi parce que dix fois moins onéreux ? Le responsable des achats d'un site peut-il être seul décisionnaire et peut-il, seul, évaluer la bonne adéquation des matières ou des prestations externalisées avec les spécifications techniques attendues ? Sur la base de quels critères choisir un prestataire de nettoyage des salles propres, un prestataire de maintenance, une société de contrôle ou de conseil en validation ? Comment est définie la règle du jeu lors des appels d'offres ? Comment sera assurée la période transitoire lors du remplacement d'un prestataire par un prestataire concurrent ?
Gestion des activités externalisées (production, essais…)
Pour toutes les activités externes, en particulier celles qui concernent l'assurance de la stérilité, il est pertinent de s'appuyer sur un cahier des charges détaillé définissant les attendus pour toute prestation ou activité externalisée en évaluant la criticité, les rôles et responsabilités de chacune des parties, et renforcé par un contrat liant client et fournisseur, ainsi qu'un document décrivant les processus d'évaluation de ces sous-traitants. Autres éléments qu'il conviendra d'évaluer : les efforts mis en œuvre par les fournisseurs lors des validations de leurs procédés.
Validation des procédés (exemple du nettoyage)
Il convient que des procédures décrivant les méthodes de nettoyage et les activités de validation contribuant à la qualité finale des produits soient en place. Elles doivent être préparées pour chaque procédé et pour chaque équipement et produit fabriqué et être en adéquation avec la charge biologique avant stérilisation et les objectifs et/ou niveaux d'assurance de stérilité définis, incluant la filtration du produit et toutes les étapes critiques du procédé de fabrication.
Validation des procédés de stérilisation
L'annexe 1 impose de nombreuses exigences lors des validations des procédés de stérilisation mais aussi pour les tests à réaliser lors des différentes étapes de qualification des équipements tels que les stérilisateurs à chaleur humide, les systèmes stérilisables en place, les fours ou les tunnels. Ainsi, une documentation décrit les efforts de validation pour tous les procédés de stérilisation et de dépyrogénation, les qualifications des équipements associés (y compris celles exécutées par les fournisseurs) et le suivi de routine par une traçabilité des cycles de production ; des règles claires et procédurées doivent être établies pour statuer sur la conformité des cycles de routine.
Maintenance préventive
Des installations et équipements de production ne peuvent rester en « état qualifié » que si les préconisations des fournisseurs quant à leur entretien et leurs maintenances préventives sont respectées. Ainsi, une description du programme de maintenance préventive est attendue pour les systèmes, les installations, les utilités et les équipements de production (salles propres également). On y inclut une description des pratiques d'étalonnage, des vérifications métrologiques et des contrôles à réaliser en cas de modifications.

- Au-delà des seuls contrôles particulaires et microbiologiques, c'est l'ensemble des paramètres démontrant la maîtrise opérationnelle des installations qui devra être pris en compte.© Aspec

- L'annexe 1 est claire sur ce point : le recours aux RABS et aux isolateurs doit être systématiquement envisagé dans le cadre de la CCS. De même que le fait de ne pas recourir à ces technologies doit y être justifié.© Berkshire Sterile Manufacturing
Nettoyage et désinfection
Les procédures opérationnelles de nettoyage (définissant les objectifs, les techniques à appliquer, les moyens mis en place pour le nettoyage des locaux et des équipements de production) doivent exister et décrire les pratiques d'entretien et de nettoyage de l'ensemble des installations, et le cas échéant des zones non classées, ainsi qu'indiquer les pratiques de désinfection des environnements classés. La validation des méthodes de nettoyage, ou tout au moins les contrôles de routine réalisés doivent être explicités. Rappelons qu'une désinfection ne peut être efficace que si un nettoyage préalable a été réalisé.
Systèmes de surveillance
Cet aspect inclut l'évaluation de la faisabilité de l'introduction de méthodes alternatives scientifiquement fondées qui optimisent la détection de la contamination environnementale.
La documentation y faisant référence doit décrire les méthodes et performances des systèmes de surveillance des particules viables et non viables, les niveaux d'alerte et d'action (justifiés et pertinents) et inclure les suivis de tendances et la gestion des écarts ou alarmes. Le suivi des tendances, les données historiques (tenues à jour) et leur analyse régulière doivent permettre la mise en place d'actions d'amélioration ainsi que l'évaluation de l'efficacité du système mis en place (a minima sur une base annuelle).
Mécanismes de prévention, analyse des tendances, enquête détaillée, détermination des causes profondes, actions correctives et préventives et outils d'investigation complets
Toutes les anomalies, les écarts, les non-conformités doivent être gérés sous « assurance qualité », présentés et perçus comme des éléments contribuant à la maîtrise des contaminations et à l'amélioration continue. Une procédure décrivant le système de gestion des actions correctives et préventives, l'analyse des tendances et les méthodes d'investigation doit exister. La traçabilité doit être assurée pour toutes les améliorations apportées aux équipements, installations, systèmes, processus, procédures et instructions ; ce qui est en général centralisé au niveau d'un logiciel informatique pour plus de fluidité.
Amélioration continue s'appuyant sur les informations dérivées de ce qui précède
Des procédures sous assurance qualité doivent s'appliquer, décrivant comment est gérée l'amélioration continue et comment est structuré et organisé le système qualité de l'entreprise. De même, la stratégie de maîtrise de la contamination doit être un document évolutif s'appuyant sur l'historique des données et la vérification périodique de l'efficacité globale du système mis en place. Entre autres, les audits internes aident à fournir les données d'entrée à exploiter pour mesurer l'efficacité du système. Si l'on se concentre, par exemple, sur la maîtrise de la contamination particulaire, le récent Technical Report ISO/TR 14644-21 souligne l'importance de la notion d'amélioration continue et la manière dont elle doit guider l'exploitation des données d'échantillonnage particulaire.
Dans ce domaine, l'application de la roue de Deming « prévoir-faire-vérifier-réagir » (PDCA) permet de comprendre l'articulation entre les résultats issus des activités de classification et de surveillance. Les changements et les améliorations conduisent à une répétition permanente de cette méthode. Le Technical Report illustre la façon dont cette dynamique influence le processus d'amélioration continue (figure 1).
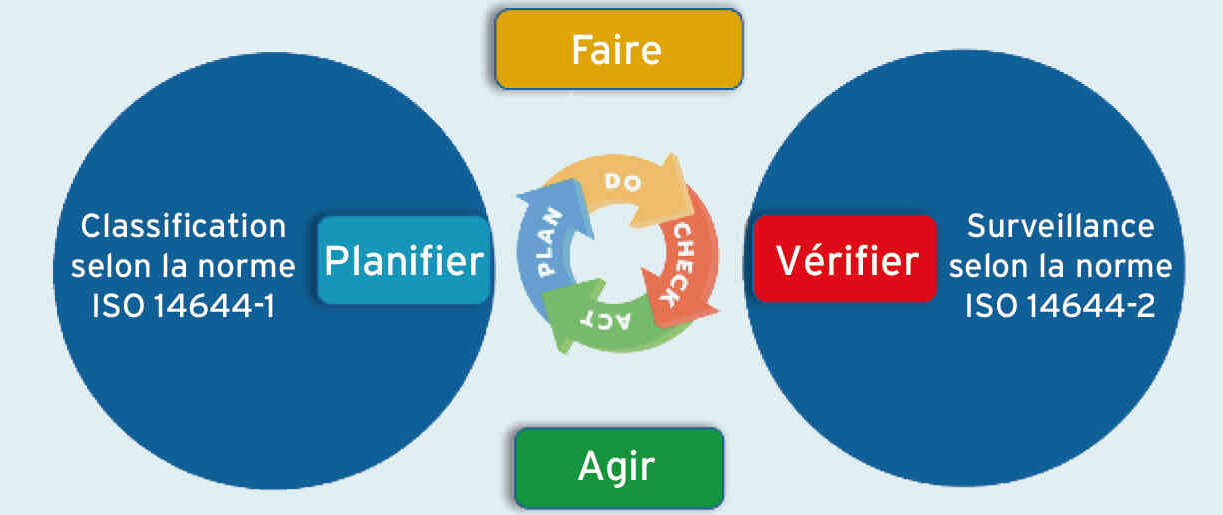
1. Stratégie de maîtrise de contamination - Pour davantage de précisions sur l'articulation entre la classification et les activités de surveillance, se reporter à l'ISO/TR 14644-21 : 2023.
Conclusions
Même si la plupart des sites et groupes industriels disposent déjà de documents décrivant leurs pratiques (soit parce que leur système qualité les a contraints à mettre en place un système documentaire avec des politiques générales, procédures, instructions, documents d'enregistrement ou autres plans directeurs de validation, protocoles parfois sans cohérence globale, soit par souci de faire progresser la qualité et de sécuriser les procédés de fabrication), il était devenu nécessaire d'élaborer un « document chapeau » permettant le lien entre tous ces documents disparates habituellement disponibles sur un site pharmaceutique.
L'exigence de rédaction et de maintien à jour par l'industriel d'une politique transversale, globale, de maîtrise et de prévention des contaminations, dite Contamination Control Strategy (CCS), est ainsi devenue réglementaire. La CCS, et également les analyses de risque, sont le fruit des compétences spécifiques et corps de métiers du site. Sachant que le personnel est « volatil » (« turn over »), il convient de tout tracer et documenter ainsi que de gérer la construction de la stratégie de maîtrise des contaminations sous assurance qualité. L'apport d'expertises externes prenant du recul, apportant un regard extérieur et challengeant les choix facilite ce type de démarche sous réserve de s'appuyer sur des personnes expérimentées et pragmatiques. Centraliser et tracer toutes les évolutions, décisions et choix de conception, de stratégie, d'approche, etc. et en donner la gestion à une équipe de pilotage multidisciplinaire interne est utile.
La complexité des méthodes d'essais, pour certains des paramètres les plus critiques tels que les prélèvements particulaires, entraîne la mobilisation d'experts pour clarifier les techniques de prélèvements et apporter la lumière sur les paramètres influant les mesures (ex. : ISO/TR 14644-21).
Sur bien des sites où la documentation est déjà présente (sans CCS rédigée au préalable), il sera nécessaire de « recoller les morceaux », créer des liens entre tous les documents participant à la maîtrise des contaminations pour rendre l'ensemble cohérent. Cet exercice consistant à mener l'analyse a posteriori, suivie de la rédaction de la CCS, est périlleux et peut conduire à conforter les pratiques déjà en place sur le site, mais également parfois à se rendre compte de défaillances, d'insuffisances ou de « trous dans la raquette », qu'il conviendra de colmater par la définition d'actions le permettant. Ceci sans oublier d'intégrer les exigences réglementaires imposées, par exemple la liste des tests et leurs périodicités, ou la nécessité de réaliser des tests d'intégrité des filtres périodiquement.






